近日李子刚/尹丰课题组在美国癌症研究会会刊《癌症研究》(Cancer Research)上在线发表题为”Stabilized peptide HDAC inhibitors derived from HDAC1 substrate H3K56 for the treatment of cancer stem-like cells in vivo” 的研究论文(DOI: 10.1158/0008-5472.CAN-18-1421),成功开发了靶向组蛋白去乙酰化酶(HDAC)的稳定多肽抑制剂,相比于已上市的药物SAHA,该多肽抑制剂表现出更好的体内抗肿瘤效果以及显著提高的安全治疗窗口。该抑制剂的研发开拓了稳定多肽在表观遗传学领域的的应用,也为研发新颖的HDAC抑制剂提供了新的方向。
通过化学手段稳定多肽的二级结构,从而提高其生物学活性是构建靶向蛋白-蛋白相互作用调控分子的重要手段之一。近年来,该课题组开发了一系列新颖的稳定多肽二级结构的方法学并将其这些方法成功地用于构建针对p53/Mdm2, 雌激素受体Alpha以及胰岛素降解酶等重要靶点的多肽调控剂。尽管稳定多肽抑制剂在靶向蛋白-蛋白相互作用中取得了一系列的重要的进展,但其在表观遗传学领域的应用则非常有限。
肿瘤的异质性和无限增殖潜力是恶性肿瘤的基本特质,肿瘤干细胞学说认为,肿瘤组织中存在的一些具有干性特征的肿瘤细胞群可能是导致肿瘤耐药性和复发的关键原因。研究表明,表观遗传学靶点,尤其是组蛋白去乙酰化酶(HDAC)在恶性肿瘤中高度表达,而且对维持肿瘤干细胞的干性至关重要,因而HDAC一直以来都是药物研发的热门靶点。FDA获批的HDAC抑制剂属于广谱抑制剂,不适用于实体瘤的治疗,在临床使用中经常出现严重的毒副作用。由于多肽对靶点有较好的亲和性以及生物相容性,该课题组首次尝试设计稳定多肽抑制剂,尝试构建能够显著提高安全治疗窗口的HDAC多肽抑制剂。
该课题组将“TD末端天冬氨酸策略”用于稳定HDAC1特异性的底物H3K56 的一段螺旋多肽,将多肽的C端H3K56位点修饰为羟肟酸官能团(该官能团为HDAC酶活抑制的经典官能团)。该多肽抑制剂对HDAC具有纳摩尔级的酶活抑制效果,在细胞水平对肿瘤干细胞(恶性卵巢癌细胞PA-1和恶性前列腺癌NTERA-2细胞)有选择性的抑制效果,而对普通的肿瘤细胞或者正常细胞基本没有毒性。在卵巢癌PA-1和前列腺癌NTERA-2的两种异位肿瘤动物模型中发现,该多肽抑制剂能达到80%的肿瘤抑制率(50mg/kg,每隔一天腹腔给药),明显高于小分子抑制剂SAHA。该多肽抑制剂的研发成功拓展了稳定多肽在表观遗传学领域的应用,该药物的高效低毒性也为靶向HDAC的抗肿瘤药物开辟了新的方向。
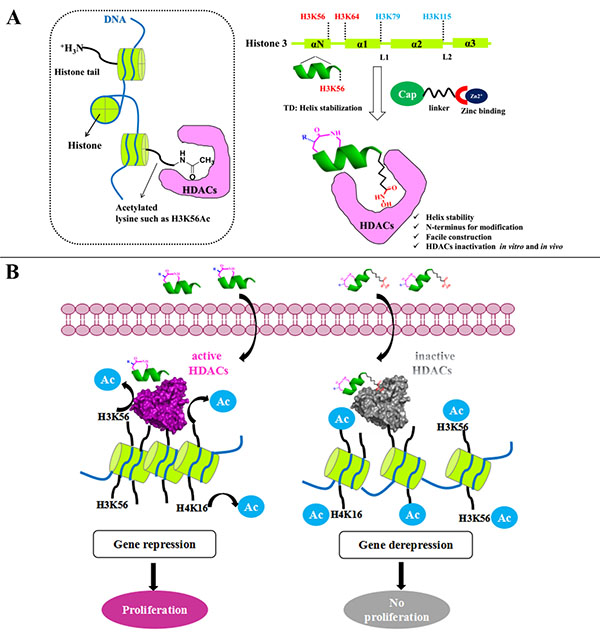
针对HDAC家族蛋白的稳定多肽药物设计
以上研究由李子刚教授和尹丰副研究员共同指导,北京大学五年级博士研究生王冬园(第一作者)以及课题组其他成员合作完成。以上工作得到了国家自然科学基金,深圳市科技创新基金以及深圳市孔雀计划项目的资助。
论文链接:http://cancerres.aacrjournals.org/content/early/2019/03/06/0008-5472.CAN-18-1421
文字:王冬园






