近日,北京大学深圳研究生院叶涛课题组完成了海洋天然产物 mandelalide B的片段合成。作者通过精心的路线设计,采用 Horner-Wadsworth-Emmons 片段组装策略以及高度立体特异性的碘诱导环化反应,构建关键的 α-羟基内酯部分,成功实现了天然产物 Mandelalide B 的四氢呋喃/ α-羟基内酯片段的立体控制合成。该工作为同家族天然产物的合成工作提供了新的研究思路。
Mandelalides A-E 和 L 是从南非阿尔哥亚湾附近的海洋海鞘 Lissoclinum sp.中分离到的一类新型大环内酯天然产物。该家族天然化合物具有24元环的内酯结构,分子中都具有2,5-顺式四氢呋喃环,2,4,6-顺式三取代四氢吡喃环,以及 Z,E 串联的共轭双烯结构。Mandelalides A 和 B 对人类 NCI-H460 肺癌细胞 (IC50 分别为 12 nM 和 29 nM) 和小鼠 Neuro-2A 神经细胞瘤 (IC50 分别为 44 nM 和 84 nM) 均有强烈的抑制活性。

图1 mandelalide家族分子结构
2014年,Fürstner 小组首次发表了对 Mandelalide A 结构推测的全合成工作。遗憾的是,该结构的核磁谱图与天然化合物的谱图无法匹配。紧随其后,叶涛教授团队发表了对 Mandelalide A 的合成研究,成功地修正了Mandelalide A 的结构,并完成其结构推测和真实结构的全合成。此后,世界范围内多个课题组参与到 Mandelalide A 的全合成研究中。Mandelalide B 的结构独特之处在于其大环骨架中镶嵌了一个 α-羟基取代的五元内酯环,使得针对Mandelalide A 的合成策略不再适用于 Mandelalide B 的全合成研究。
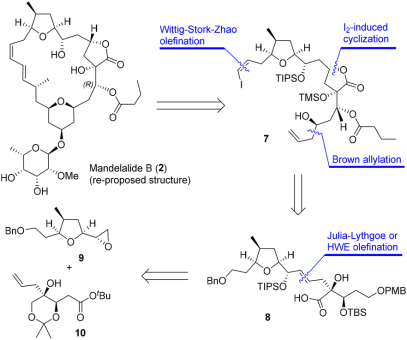
图2 mandelalide B的逆合成分析
近日,北京大学深圳研究生院叶涛教授团队针对 Mandelalide B 提出了新型的合成策略,并完成了目标分子中四氢呋喃/ α-羟基内酯片段 (结构 7) 的合成工作。逆合成分析如图所示,片段7的合成可通过碘诱导不对称内酯化反应构建α-羟基内酯环,采用 Wittig-Stork-Zhao 烯化反应构建顺式烯基碘活性位点,采用布朗不对称烯丙基化反应构建端位高烯丙醇结构。羧酸8 可以简单片段9和10为原料,通过 Julia-Lythgoe 反应或 Horner-Wadsworth-Emmons (HWE) 反应连接得到。
作者首先尝试了 Julia-Lythgoe 片段连接策略,但遗憾地失败了。随后,成功通过 HWE 反应实现了片段连接。该工作中,作者分别合成了膦酸酯16和醛13,通过条件筛查,选择了改良版 Paterson 反应条件,成功制备出烯酮化合物17,且未观察到 Z-式产物。化合物17经过不对称 Corey-Bakshi-Shibata 还原反应 (dr > 20:1) 及保护基调整,制备出二醇化合物 22。化合物 22经过 Dess-Martin 氧化和 Pinnick 氧化反应,转化为羧酸化合物 8 (关环前体)。在四异丙氧钛和碘的作用下,化合物 8顺利实现立体专一性的内酯化反应 (dr > 20:1),再经自由基脱碘反应,转化为大片段化合物 25。
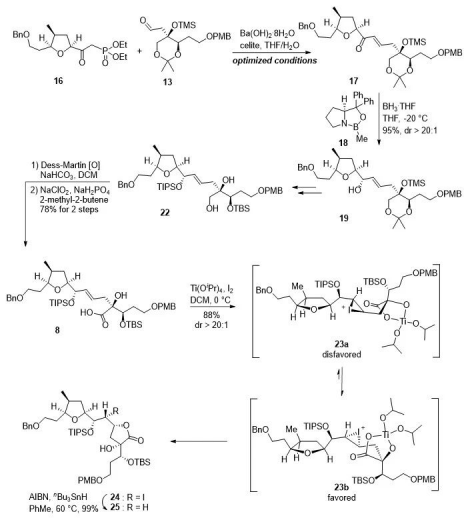
图3 片段25的合成
化合物 25两端的活性基团均可参与进一步的转化反应。作者首先将化合物 25的 PMB 醚转化为高烯丙醇结构,然而在后续脱苄醚的过程中无法得到目标产物。因此,作者调整了合成方案。如图4所示,新方案首先采用雷尼镍催化氢化选择性脱除苄醚,随后将所得醇转化为烯基碘化合物35。化合物35经过简单官能团转化及丁酰基化,再脱除 PMB 醚后转化为 37。化合物37通过 Dess-Martin 氧化为醛后,经过布朗不对称烯丙基化反应,制备出目标四氢呋喃/ α-羟基内酯片段 7。
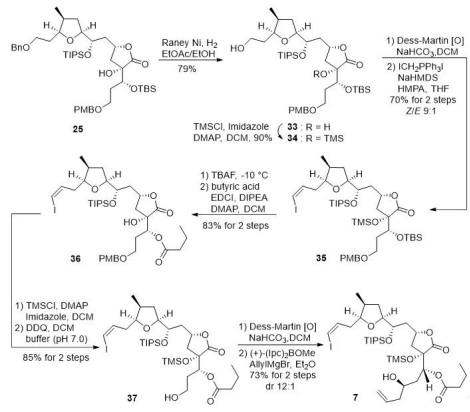

图4 目标片段7的合成
闫加磊博士为本文的第一作者,该研究得到了国家自然科学基金和深圳市高校可持续发展支持基金的支持。
原文链接:https://pubs.rsc.org/en/content/articlelanding/2024/qo/d4qo01433b






