研究进展
近期,北京大学化学生物学与生物技术学院、科学智能学院陈语谦课题组与新加坡国立大学合作,开发了一种基于高效等变模型的机器学习原子间势模型E2GNN,不同于传统等变模型依赖复杂的高阶张量表示,E2GNN 采用标量-向量的简洁表示对等变特征进行编码,并结合全局消息传递机制,更精准地描述了长程原子间相互作用,实现高效且物理合理的势能预测。相关成果以“Efficient equivariant model for machine learning interatomic potentials”为题,发表于AI4M领域著名期刊npj Computational Materials(中科院一区Top),并入选Featured Article。

精准且高效地预测原子间势能是推进材料设计与模拟的核心环节。然而,现有深度学习方法虽在加速计算方面展现出巨大潜力,却常难以同时兼顾预测精度与计算效率,尤其在大规模体系模拟中挑战尤为突出。要实现“又快又准”的势能预测,需要兼顾两个要点:其一,为保持物理合理性,模型输出的能量须对刚体变换(平移、旋转)不变,预测的力场则需对平移不变、对旋转等变,以确保物理一致性;其二,模型设计在满足上述等变性约束的同时,还应维持足够的计算效率,以适应大规模模拟的需求。
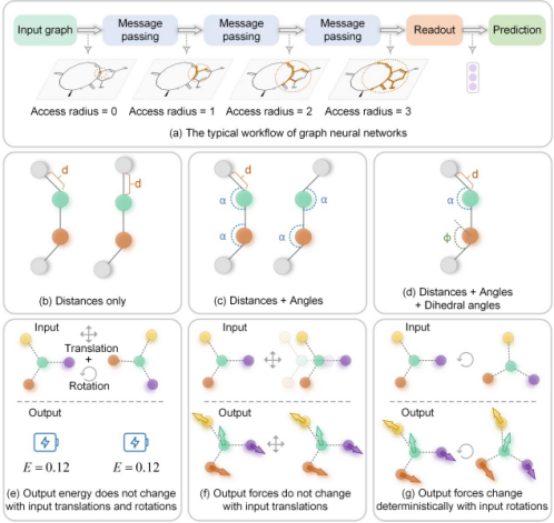
图1 图神经网络的基本原理以及等变与不变概念的解释
为解决上述难题,研究团队提出了一种兼具精度与效率的等变图神经网络(E2GNN)。与传统等变模型依赖复杂高阶张量表示不同,E2GNN 采用简洁的“标量-向量”混合表示编码等变特征,并引入全局消息传递机制,以精确捕捉长程原子相互作用,从而实现高精度、高效率的势能预测。全局消息传递与门控消息传递机制的协同,又进一步提升了模型整体表现。

图2 E2GNN的原理图
实验结果表明,在催化剂、分子体系和有机异构体等多个数据集中,E2GNN 均优于当前代表性基线模型,同时在计算效率上展现出明显优势。此外,基于 E2GNN 力场进行的固态、液态和气态体系的分子动力学模拟,其预测精度可达到第一性原理级别。本研究提出的高精度、高效率原子间势能预测模型,有望加速新材料发现、优化现有材料性能,并为材料设计提供更高效的数据驱动工具,助力材料科学计算迈向更精准、更高效的新阶段。
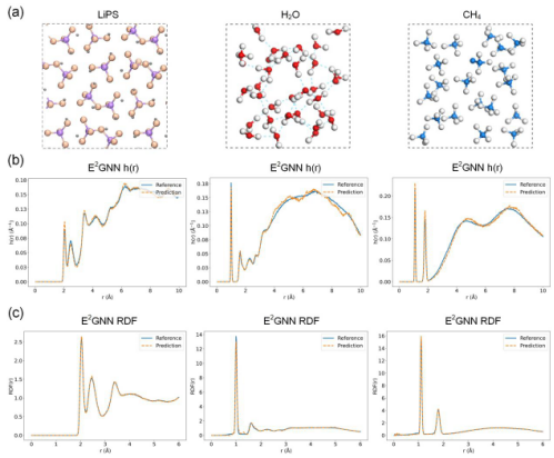
图3 对固体、液体和气体分子动力学模拟
论文链接
https://www.nature.com/articles/s41524-025-01535-3






